UDI – Unique Device Identifier

Der Unique Device Identifier (UDI) dient der eindeutigen Kennzeichnung der Produkte. Mit Geltungsbeginn der europäischen Verordnungen für Medizinprodukte und In-vitro-Diagnostika (MDR und IVDR) ist eine Registrierung des UDI und der dazugehörigen Produktinformationen in der Europäischen Datenbank EUDAMED verpflichtend. Die UDI soll als maschinenlesbare Kennzeichen (z.B. Barcode) und in Klarschrift auf dem Produkt und der Verpackung […]
Medizinproduktebetreiber-Verordnung MPBetreibV

Die Medizinprodukte-Betreiberverordnung richtet sich an die (professionellen) Betreiber und Anwender von Medizinprodukten. Betreiber eines Medizinproduktes ist jede natürliche oder juristische Person, die für den Betrieb der Gesundheitseinrichtung verantwortlich ist, in der das Medizinprodukt durch dessen Beschäftigte betrieben oder angewendet wird.Abweichend von Satz 1 ist Betreiber eines Medizinproduktes, das im Besitz eines Angehörigen der Heilberufe oder […]
Klassifizierung von Medizinprodukten nach MDR
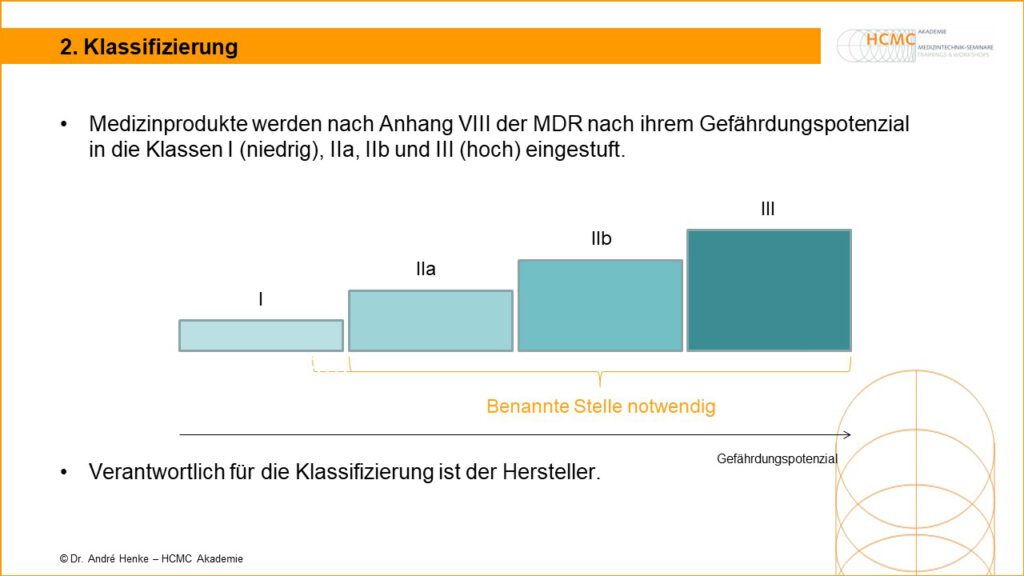
Klassifizierung von Medizinprodukten Medizinprodukte werden nach Anhang VIII der MDR nach ihrem Gefährdungspotenzialin die Klassen I (niedrig), IIa, IIb und III (hoch) eingestuft. Verantwortlich für die Klassifizierung ist der Hersteller. Mehr dazu in unseren Seminaren zum Medizinprodukteberater.
Klassifizierung IVDs nach IVDR Artikel 39 und Anhang VIII (neue IVDR)
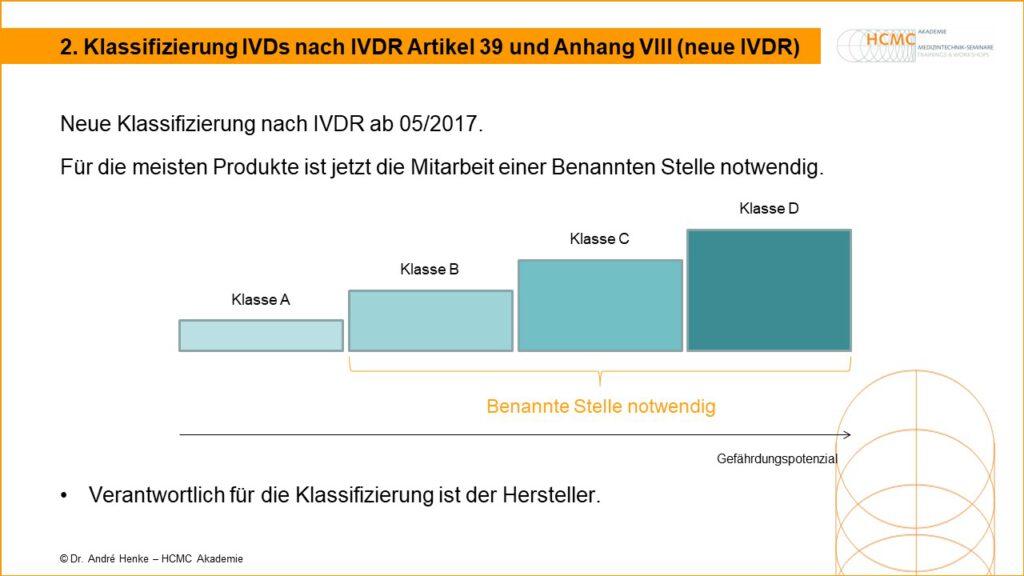
Neue Klassifizierung nach IVDR ab 05/2017. Für die meisten Produkte ist jetzt die Mitarbeit einer Benannten Stelle notwendig. Verantwortlich für die Klassifizierung ist der Hersteller. Mehr dazu in unseren Seminaren zum Medizinprodukteberater.
Klinische Bewertung

Für jedes in Europa auf den Markt gebrachtes Medizinprodukt ist eine klinische Bewertung durchzuführen. Der Hersteller muss den Zweck des Medizinproduktes angeben, die Erfüllung der grundlegenden Anforderungen bezüglich Sicherheit und Leistungsfähigkeit nachweisen sowie die unerwünschten Nebenwirkungen bei bestimmungsgemäßem Gebrauch beurteilen. Dabei erfolgt die Bewertung jedes Medizinprodukts anhand von klinischen Daten aus der wissenschaftlichen Literatur oder […]
Klinische Prüfung

Liegen keine ausreichenden klinischen Daten vor, so muss der Hersteller eine klinische Prüfung durchführen (DIN EN ISO 14155 Teil 1 und 2). Der Umfang der klinischen Prüfung liegt in der Verantwortung des Herstellers. Für eine klinische Prüfung ist die Genehmigung durch das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) oder das Paul-Ehrlich-Institut (PEI), zuständig für Hochrisiko-In-vitro-Diagnostika, […]
Konformitätsbewertung
Mit der Konformitätsbewertung erklärt der Hersteller, dass sein Medizinprodukt mit den grundlegenden Anforderungen der relevanten EU-Richtlinie / der MDR/IVDR übereinstimmt. Das Konformitätsbewertungsverfahren ist abhängig von der Klassifizierung der Medizinprodukte. Die Konformitätsbewertungsverfahren sind im Artikel 52 MDR und den Anhängen IX – XI beschrieben. Mehr dazu in unseren Seminaren zum Medizinprodukteberater.
Technische Dokumentation

Die technische Dokumentation wird je nach dem gewählten Konformitätsbewertungsverfahren erstellt. Sie enthält u. a. Produktbeschreibung, Konstruktions- und Fertigungszeichnungen, Risikoanalyse (vgl. auch DIN EN ISO 14971), Prüfberichte, Liste der angewandten Normen, Gebrauchsanweisung und Kennzeichnung. Die technische Dokumentation muss mindestens 5 Jahre, im Falle von implantierbaren Produkten für mindestens 15 Jahre nach Herstellung des letzten Produkts zur […]
Gebrauchsanweisung

Kapitel III MDR (Anforderungen an die mit dem Produkt gelieferten Informationen) Jedem Produkt werden die notwendigen Angaben beigefügt, Mehr dazu in unseren Seminaren zum Medizinprodukteberater.
Elektronische Gebrauchsanweisung für Medizinprodukte (eIFU)

Neu: Durchführungsverordnung (EU) 2021/2226 für MDR-Medizinprodukte (nicht MDD), möglich wenn: Mehr dazu in unseren Seminaren zum Medizinprodukteberater.